For Intravenous Use Only
INDICATION AND USAGE
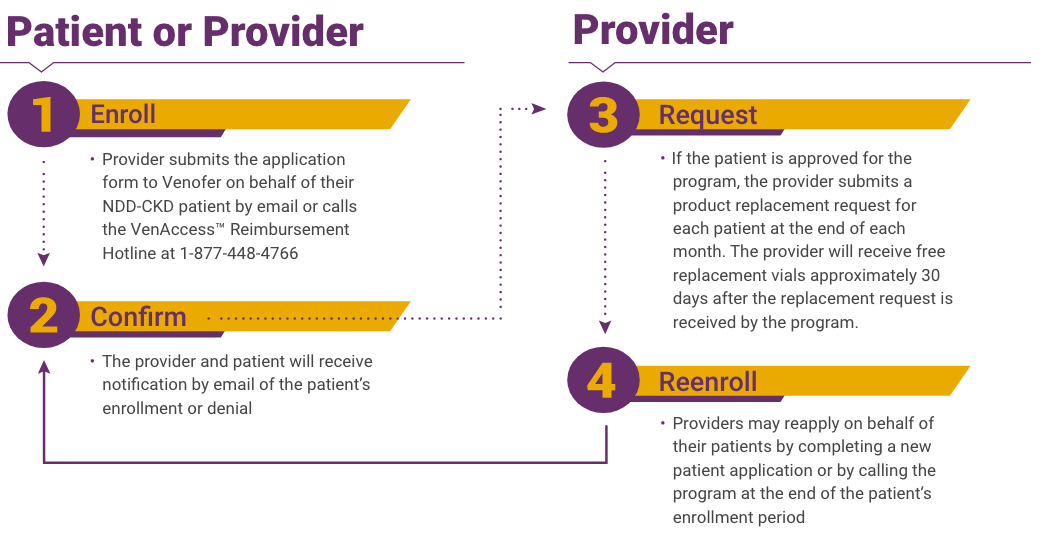
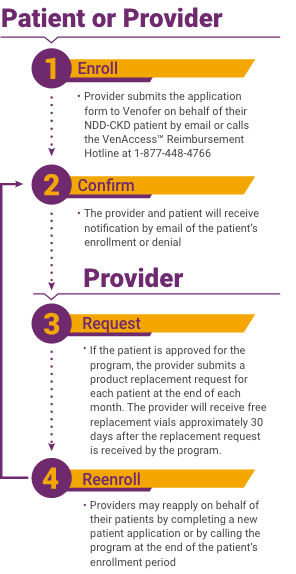
Venofer® (iron sucrose) injection, USP is indicated for the treatment of iron deficiency anemia (IDA) in patients with chronic kidney disease (CKD).
IMPORTANT SAFETY INFORMATION
DOSAGE AND ADMINISTRATION
Pediatric Patients (2 Years of Age and Older)
The dosing for iron replacement treatment in pediatric patients with Peritoneal or Hemodialysis-Dependent - CKD or Non-Dialysis Dependent CKD have not been established.
CONTRAINDICATIONS
Known hypersensitivity to Venofer.
WARNINGS AND PRECAUTIONS
Serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening and fatal, have been reported in patients receiving Venofer. Patients may present with shock, clinically significant hypotension, loss of consciousness and/or collapse. If hypersensitivity reactions or signs of intolerance occur during administration, stop Venofer immediately. Monitor patients for signs and symptoms of hypersensitivity during and after Venofer administration for at least 30 minutes and until clinically stable following completion of the infusion. Only administer Venofer when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions. Most reactions associated with intravenous iron preparations occur within 30 minutes of the completion of the infusion.
Venofer may cause clinically significant hypotension. Monitor for signs and symptoms of hypotension following each administration of Venofer. Hypotension following administration of Venofer may be related to rate of administration and/or total dose delivered.
Excessive therapy with parenteral iron can lead to excess storage of iron with the possibility of iatrogenic hemosiderosis. All adult and pediatric patients receiving Venofer require periodic monitoring of hematologic and iron parameters (hemoglobin, hematocrit, serum ferritin and transferrin saturation). Do not administer Venofer to patients with evidence of iron overload. Transferrin saturation (TSAT) values increase rapidly after intravenous administration of iron sucrose; do not perform serum iron measurements for at least 48 hours after intravenous dosing.
ADVERSE REACTIONS
Adult Patients: The most common adverse reactions (≥2%) include diarrhea, nausea, vomiting, headache, dizziness, hypotension, pruritus, pain in extremity, arthralgia, back pain, muscle cramp, injection site reactions, chest pain and peripheral edema.
Pediatric Patients: The most common adverse reactions (≥2%) are headache, respiratory tract viral infection, peritonitis, vomiting, pyrexia, dizziness, cough, nausea, arteriovenous fistula thrombosis, hypotension and hypertension.
Post-Marketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. In post-marketing safety studies of Venofer in 1,051 patients with HDD-CKD, adverse reactions reported by >1% were cardiac failure congestive, sepsis and dysgeusia.
- Immune system disorders: anaphylactic-type reactions, angioedema
- Psychiatric disorders: confusion
- Nervous system disorders: convulsions, collapse, light-headedness, loss-of-consciousness
- Cardiovascular system: bradycardia, shock, acute myocardial ischemia with or without myocardial infarction or with in-stent thrombosis in the context of a hypersensitivity reaction
- Respiratory, thoracic and mediastinal disorders: bronchospasm, dyspnea
- Musculoskeletal and connective tissue disorders: back pain, swelling of the joints
- Renal and urinary disorders: chromaturia
- General disorders and administration site conditions: hyperhidrosis
Symptoms associated with Venofer total dosage or infusing too rapidly included hypotension, dyspnea, headache, vomiting, nausea, dizziness, joint aches, paresthesia, abdominal and muscle pain, edema and cardiovascular collapse. These adverse reactions have occurred up to 30 minutes after the administration of Venofer injection. Reactions have occurred following the first dose or subsequent doses of Venofer. Slowing the infusion rate may alleviate symptoms.
Injection site discoloration has been reported following extravasation. Assure stable intravenous access to avoid extravasation.
DRUG INTERACTIONS
Venofer may reduce the absorption of concomitantly administered oral iron preparations.
USE IN SPECIFIC POPULATIONS
Pregnancy: Risk Summary-Clinical Considerations
Untreated IDA in pregnancy is associated with adverse maternal outcomes such as post-partum anemia. Adverse pregnancy outcomes associated with IDA include increased risk for preterm delivery and low birth weight.
Severe adverse reactions including circulatory failure (severe hypotension, shock including in the context of anaphylactic reaction) may occur in pregnant women with parenteral iron products (such as Venofer) which may cause fetal bradycardia, especially during the second and third trimester.
Geriatric Use
Dose administration to an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
For additional Safety Information, please see Full Prescribing Information.
You are encouraged to report Adverse Drug Events to American Regent, Inc. at 1-800-734-9236 or to the FDA by visiting www.fda.gov/medwatch or calling 1-800-FDA-1088.
REF-0262 6/2022